Blog Posts
Use of position restraint in Molecular Dynamics Simulation
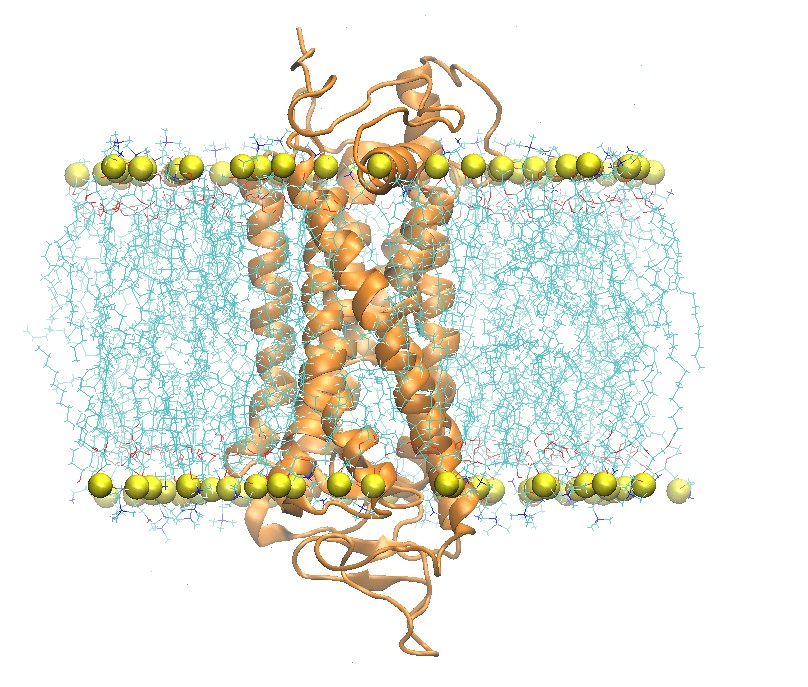
I mostly work with transmembrane protein which is such an interesting system to simulate. The idea behind the position restraints is very much centered around equilibrating a transmembrane protein, although it should also be applied to soluble/globular proteins (protein in water).
The very first simulation we perform will have velocities that are generated for each atom from something known as a maxwell boltzmann distribution (BD). A BD is a simple function which can be used to define particle speed according to the temperature of a system and with it being a probability distribution functions every atom gets a velocity from somewhere over this distribution. Most of the atoms will get a velocity somewhere close to the mean of the BD, but some e.g., those more than 1.5 standard deviations from the mean, will get very high or very low velocities.
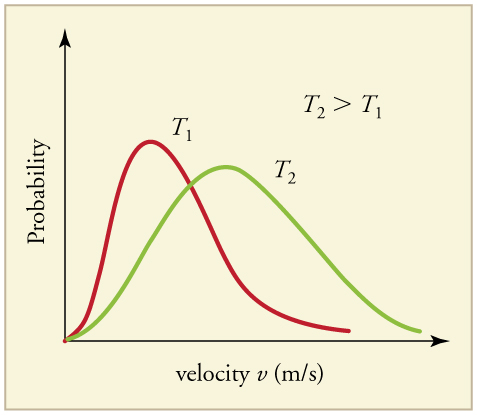
The molecular dynamics integrator uses these velocities to predict the position of the atoms and associated forces. Therefore, if your system contains very large velocities we will have particles that will move a lot further than a particle with a velocity close to the mean from the BD. Large atom movements (associated with high velocities) normally produces large particle-particle forces and thus causing a chain of large particle movements across your system. It is very easy for secondary structures e.g., beta-sheet and alpha helix to change if there are large velocities in neighbouring atoms. The longer we run your system (equilibration time) the more likely that these velocities eventually smooth out to be similar to the mean velocity associated with the BD at your system temperature. You can apply position restraints on important parts of the system e.g., the crystallographic shape of a protein whilst any high velocities gradually smooth out.
This is why we have position restraints on a protein during equilibration: to provide sufficient time for high and low particle velocities to reach the mean Boltzmann distribution particle speed whilst safe-guarding against the protein from unfolding due to initial high velocities.